






FDA approval process — Bringing a medical device or healthcare product to the US market entails more than only speed or innovation. With written proof, you are showing that your product is safe, secure, and sufficiently dependable for actual patient usage. One of the most stringent rules worldwide, the US Food and Drug Administration does not approve goods depending on intent, vision, or advertising assertions. It approves them according to verifiable testing, risk mitigation measures, and compliance records.
Approvals can be postponed for months or even years by even one misplaced document, an ambiguous planned use statement, or poor cybersecurity validation. Many businesses only find these holes after submission, when answering FDA questions costs time, stress, and money. Teams are compelled to use reactive solutions at that point rather than managed preparation.
That helps you to see why knowing the process before filing is so important. This guide covers the whole medical device FDA approval process, how FDA medical device clearance really works, what reviewers thoroughly assess, and how companies might cut down on delays by properly fulfilling the requirements for FDA approval from day one.
FDA Approval Process: What Is It?
The FDA clearance process is the official regulatory structure established by the US Food and Drug Administration to ensure that medical products entering the US market meet high standards for safety, effectiveness, and compliance. This method assesses far more than just whether medical equipment operates as intended in a demonstration setting.
FDA reviewers study the performance of a device in actual clinical settings, including failure scenarios and edge cases. They evaluate how hazards are found, noted, reduced, and monitored over time. This covers software behavior, data flow, access restriction, cybersecurity measures, and hardware reliability, especially for network-connected or software-driven devices.
Depending on the type of medical device, planned clinical use, degree of patient risk, and connectivity, the medical device FDA clearance process differs greatly. Compared with a cloud-linked diagnostic system, a standalone mechanical tool receives far less examination. Early knowledge of these differences spares unneeded redesigns, multiple filings, and regulatory backtracking.
👉 Discover: Medical Device Cybersecurity: The Importance In Healthcare
Why The FDA Approval Mechanism Is So Important
The FDA’s approval process is not a paperwork ritual. It is a legal and regulatory protection meant to shield healthcare systems, medical practitioners, and patients from dangerous or useless medical devices. Legal marketing, sales, distribution, or even advertisement of a medical device in the United States without FDA clearance is not possible. This holds for linked digital health products, software-based medical devices, and hardware devices as well.
Companies that do not grasp FDA expectations often have serious repercussions. These include FDA warning letters, import alerts, forced product recalls, civil fines, and, in some cases, complete market withdrawal. Once a firm shows up in FDA enforcement data, regaining confidence among partners, clients, and regulators gets much harder.
Commercially, FDA medical device clearance has a direct effect on revenue potential and adoption rate. Hospitals, medical professionals, and insurance companies depend on FDA approval as proof of an independent product safety and effectiveness assessment. Many procurement and compliance departments regard FDA permission as a nonnegotiable condition before any vendor onboarding or preliminary implementation.
Reimbursement and insurance coverage are also impacted by FDA clearance. Many public and private payers want FDA clearance before approving reimbursements. Lack of coverage might cause even clinically useful devices to fall financially without it.
FDA clearance lowers long-term operating risk beyond fast market access. It lessens legal exposure, defines liability limits, and offers written evidence of risk assessment and reduction following regulatory criteria. This turns out to be very important in audits, lawsuits, or negative event inquiries.
Often, acting as a regulatory baseline worldwide is FDA clearance. Many foreign regulatory agencies cite FDA requirements in their assessment of foreign devices. For businesses thinking of global growth, the FDA approval procedure transforms into a foundation instead of a destination.
Explaining FDA Medical Device Categorization
Manufacturers must determine how the FDA categorises their medical device before initiating the approval process. The degree of regulatory control used affects testing depth, documentation demands, review deadlines, and post-market obligations—that is, device classification.
The FDA classifies based on risk. The more evidence the FDA needs to show safety and efficacy, the higher the possible risk to patients or consumers. Early misinterpretation of categorization results in inaccurate submissions, lost development time, and expensive revisions.
Class I Devices (Low Risk)
Bandages, manual surgical instruments, and basic medical supplies are among the Class I devices. Normally devoid of software or communication, these gadgets provide little danger when properly used.
They fall under overall regulations, including precise labeling, manufacturing quality systems, complaint resolution, and record keeping. Although most Class I devices are exempt from premarket applications, producers are still responsible for ensuring compliance and inspection readiness.
Failure to abide by common controls might yet lead to enforcement action even when a premarket submission is not necessary.
Class II Devices (Moderate Risk)
Many software-driven medical devices, as well as infusion pumps, patient monitoring systems, and diagnostic imaging equipment, are included among Class II devices. Though they usually interact with patient data or clinical processes, these products present little danger.
They are under particular checks, including performance standards, software validation, cybersecurity safeguards, and post-market surveillance. Most Class II devices follow the 510(k) process and must prove great similarity to a legally sold predicate device.
For software-enabled Class II devices, cybersecurity proof has become a top priority during FDA review.
Class III Devices (High Risk)
Pacemakers, implantable defibrillators, and life-sustaining implants are all classified under Class III devices. These gadgets have the highest patient risk if they stop or support life.
They need Premarket Approval (PMA), which includes clinical trials, thorough risk analysis, long-term safety data, and the most stringent FDA review cycle. PMA submissions usually take years to complete and call for ongoing engagement with the FDA.
For Class II and Class III devices that rely on software or connectivity, independent security testing strengthens the justification for classification and submission confidence. Here is how Qualysec enhances this confidence: Book a free consultation to learn more!
Requirements For FDA Approval: You Must Not Miss
Meeting the criteria for FDA approval requires much more than just filing papers. FDA assesses the management of risks throughout the life cycle of the product.
This covers secure development techniques, vulnerability management procedures, incident response preparation, system updates, and post-market monitoring. Connected devices have especially severe expectations.
FDA increasingly looks for evidence that approved security governance continues. One-time testing is now inadequate.
Independent validation lowers regulatory pushback and strengthens claims of compliance.
Categories Of FDA Medical Device Submissions
Selecting the right submission route is a key element of FDA medical device approval. One of the most frequent causes for slowed evaluations, refusal letters, and repeated applications is choosing the incorrect route.
510(k) Premarket Notification
When a device is almost identical to a legally sold predicate device, the 510(k) route is followed. Substantive equivalence means the same intended use and comparable technical features.
Manufacturers must provide software validation evidence, risk management papers, performance test results, and increasingly cybersecurity controls. Though quicker than PMA, 510(k) submissions are sometimes held up because of missing papers.
De Novo Classification
The de novo route kicks in when no prior gadget is available, yet overall risk remains modest. Typically employed for creative gadgets that add new capability without great patient risk.
Often, defining fresh device categories that the following makers might cite, successful de novo submissions help. Nevertheless, De Novo submissions need substantial safety, performance, and risk reduction proof.
Premarket Approval (PMA)
Class III devices need PMA, which is the most rigorous review route. It calls for clinical trials, long-term safety and efficacy results, cybersecurity validation, and plenty of documentation.
The PMA review cycles of the FDA are extended and comprise many rounds of clarification and questioning.
Investigational Device Exemption (IDE)
Before complete approval, an IDE lets a gadget be used in clinical trials. The FDA monitors IDE studies to safeguard patient safety and guarantee ethical compliance.
Identify and fix cybersecurity gaps before FDA review – partner with Qualysec.
FDA Submission Pathways Vs Evidence Expectations
| Submission Type | Risk Level | Key Evidence Required | Typical Review Time |
| 510(k) | Moderate | Predicate comparison, performance testing, risk analysis | 3–6 months |
| De Novo | Moderate | Safety data, risk controls, clinical rationale | 6–9 months |
| PMA | High | Clinical trials, full risk management, security validation | 12–24 months |
| IDE | Research | Study protocol, patient safety controls | Ongoing |
Get your medical device FDA-ready with comprehensive penetration testing.
Step-by-Step FDA Approval Process
FDA approvals are not a one-time submission operation. Starting early in product planning, it is an orderly cycle that stretches far beyond market entry. Many companies fail because they view FDA approval as a paperwork exercise rather than a risk- and evidence-driven process. Every level relies on the one before it; jumping or hurrying any stage increases the likelihood of denial, delays, or post-market enforcement.
The medical equipment FDA authorization process calls for coordination of regulatory documents, software behavior, engineering design, clinical objectives, and security precautions. Uniformity in every submitted artifact is expected by FDA reviewers. Should there be inconsistencies, they assume the underlying risk is either not completely understood or under control.
Depth will let you understand not only what to do but also why the FDA demands it via the true step-by-step breakdown below.
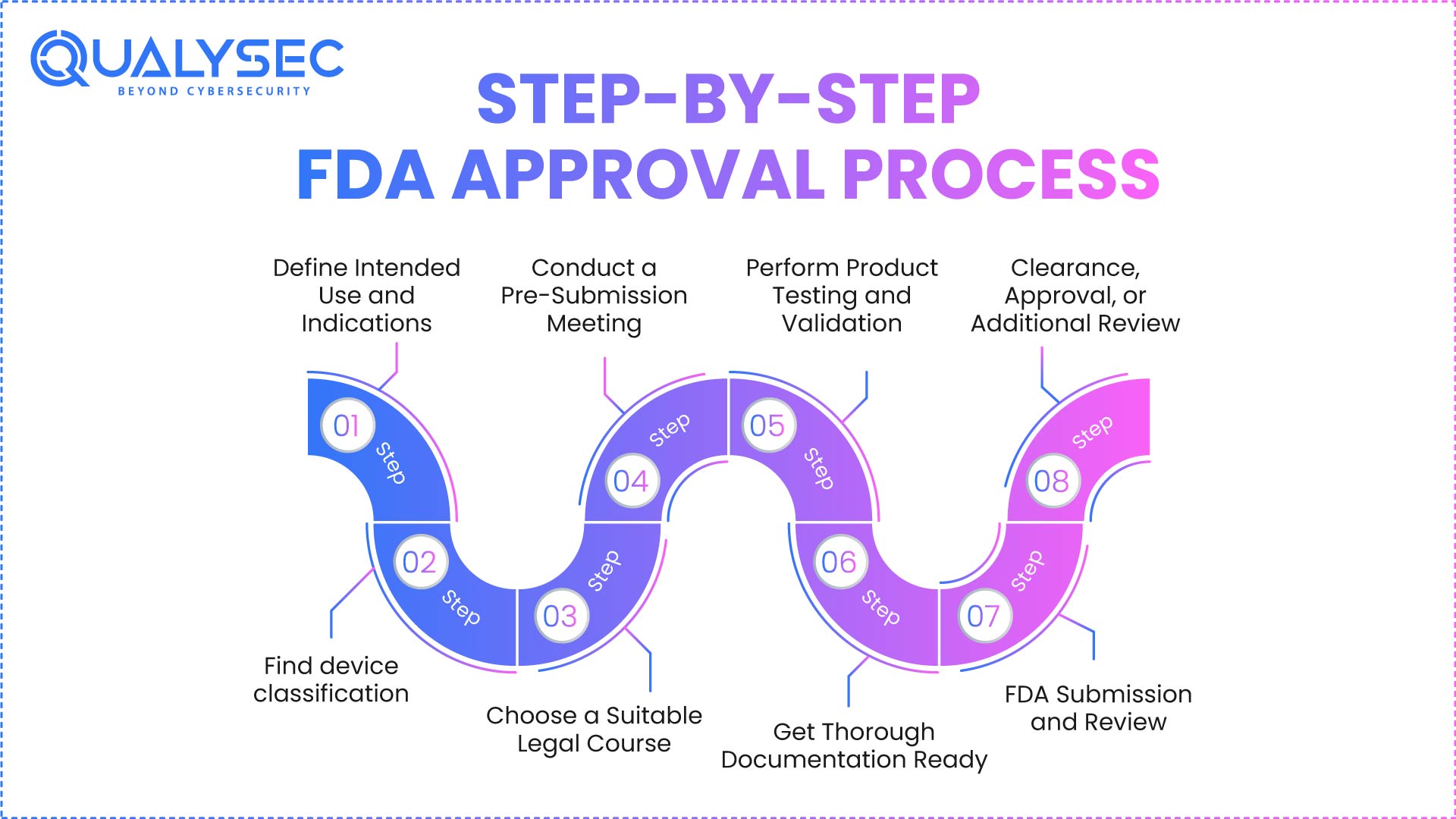
Step 1: Define Intended Use and Indications
This stage underpins the whole FDA approval process. The intended use defines exactly who the device is made for, what it does, and in which clinical setting it operates. FDA examiners mostly rely on statements of intended use to determine risk, categorize goods, and determine the sufficiency of the supplied data.
Labels, technical manuals, risk management logs, software descriptions, and FDA submission documents all need to have constant intended use terminology. Even small modifications in wording cause rejections, more review cycles, or questions for clarification. Inconsistent language is seen by the FDA as evidence that the manufacturer does not completely grasp the clinical constraints of the goods.
The breadth of testing is also influenced by unambiguous definitions of intended use. A building designed solely for monitoring will be under a different level of scrutiny than one built for diagnostic purposes. Claims of treatment, prediction, or decision support quickly increase regulatory expectations.
Usually, poorly described intended use leads to incorrect device classification, wrong submission paths, and late-stage redesigns. Following submission, changing the intended use is rather challenging and normally calls for resubmission.
Companies that spend time here lower risk throughout the FDA medical device approval procedure.
Step 2: Find device classification
Your product is classified as Class I, Class II, or Class III based on the device. This choice directly impacts regulatory burden, testing depth, documentation quantity, and review timelines under the medical device FDA approval process.
Based on patient risk, not technological complexity, the FDA categorizes devices. More strict classification may be required for a fundamental instrument that directly influences patient care than for a sophisticated technical device mostly employed for data display.
One of the main reasons for rejection is misclassification. Filing a 510(k) often fails when a PMA is needed. The FDA wants producers to explain their classification using the released guidance, databases, and predicate analysis.
Early validation of categorization enables groups to plan resource allocation, schedules, and testing budgets correctly. Furthermore, it guarantees alignment among engineering, medical, and regulatory departments.
Correct categorization is not negotiable. In the FDA approval process, it acts as a gateway measure.
Step 3: Choose a Suitable Legal Course
Once classification is established, producers must choose the appropriate regulatory channel: 510(k), De Novo, PMA, or IDE. This decision affects the FDA’s whole approval plan for medical equipment.
Every path has unique requirements for FDA clearance, review scope, and evidential standards. A 510(k) highlights great equivalency even though PMA seeks original clinical data and long-term safety information.
Choosing the incorrect route leads to lost time, unnecessary documents, and pointless testing. Without some delay, the FDA won’t turn in a submission for you. The producer is held responsible.
Decisions on strategic routes also have an impact on pricing. PMA submissions are pricey and drawn out; De Novo could help to lessen the load for fresh but intermediate-risk instruments.
Skilled producers match route choices to legal objectives as well as commercial ones.
Step 4: Conduct a Pre-Submission Meeting
Early contact with FDA reviewers is made possible by a pre-submission meeting, therefore allowing early contact before the official filing. Though not compulsory, this stage is among the most efficient FDA-approved devices.
During these meetings, producers may verify categories, review testing timelines, and change paperwork standards. At this phase, FDA remarks often save months of future work.
Pre-submissions especially help networked or software-driven devices since cybersecurity expectations might differ depending on design and use.
Though it gives direction, the FDA rejects goods during pre-submission. Seeing this opportunity raises instability and risk.
Step 5: Perform Product Testing and Validation
The FDA demands comprehensive technical proof of dependability, efficacy, and safety. This includes software validation, performance validation, functional testing, and cybersecurity risk assessment.
For software-driven or linked devices, the FDA’s medical device approval now calls for basic cybersecurity. The FDA looks over time at how errors are discovered, reduced, and tracked.
Rather than ideal laboratory settings, testing has to mirror actual world circumstances. Generally rejecting proof of poor risk management, FDA reviewers
Many manufacturers utilize outside security companies such as Qualysec to carry out penetration tests and application security verification, matching FDA criteria. Early verification helps to prevent problems later in life.



























































































































































































































































































































































































































































































































































































































0 Comments